GWASČŦŧųŌō―MęP·ÖÎöĩÚŌŧÆÚĢšĩþž°ļņĘ―ÞDQ
GWASĢĻČŦŧųŌō―MęPÂŅÐūŋĢŽGenome-Wide Association StudyĢĐĘĮŌŧ·NÍĻß^ßÃčČŦŧųŌō―M·ķúČĩÄßzũŨŪĢĻČįΚËÜÕËáķāBÐÔĢŽSNPĢĐĢŽĪÕŌÅcĖØķĻÐÔ îŧōžēēĄï@ÖøęPÂĩÄŧųŌōÎŧücĩÄ·―·ĻĄĢ
ÔÚßMÐÐGWAS·ÖÎöÖŪĮ°ĢŽÐčŌŠßxņšÏßmĩÄČšówĢŽßMÐÐĩþĘÕžŊĄĢëHđĪŨũÖаlŽFÓÐÐĐŋÍôļųąūēŧÖŠĩĀGWAS·ÖÎöÐčŌŠĘēÃīĩþĢŽĩĒÕ`·ÖÎößMķČĢŽÄĮÐčŌŠĘÕžŊÄÄÐĐĩþÄØĢŋÖũŌŠÐčŌŠÉĩþĢšąíÐÍĩþšÍŧųŌōÐÍĩþĄĢ
01 ąíÐÍĩþ
ÕÆðąíÐÍĩþĢŽÎŌÏČíÁË―âÏÂGWASĩÄąíÐÍÐÔ îĄĢGWASÖÐĩÄąíÐÍÐÔ îŋÉŌÔ·ÖéŌÔÏÂČýîĢš
ĒŲ ĩÁŋÐÔ îĢĻQuantitative TraitsĢĐĢšĘĮÖļŋÉŌÔÓÃĩŨÖÖĩíÃčĘöĩÄÐÔ îĄĢąČČįÉíļßĢĻcmĢĐ/ówÖØĢĻkgĢĐ/ŨŅÁĢĩĢĻĢĐ/ŪaÁŋĢĻkg/ŪĢĐĢŽ·ÖÎörąíÐÍÖą―ÓĘđÓÃūßówĩÖĩĢŧ
ĒÚ Ų|ÁŋÐÔ îĢĻQualitative TraitsĢĐĢšÅcĩÁŋÐÔ îÏā·īĢŽÆäo·ĻÓÃđĖķĻĩÖĩąíĘūĢŽķøĘĮąíŽFģöŌŧ·N îBĄĢąČČįŧĻÉŦĢĻžtĄĒüSĄĒ°ŨĩČĢĐ/đûÐÎ îĢĻAĄĒEAĩČĢĐ/žēēĄĢĻÓÐŧōoĢĐĢŽąíÐÍŋÉŌÔÓÃĩÖĩšŧŊąíĘūĢŧ
ĒÛ ·ÖžÐÔ îĢĻOrdinal TraitsĢĐĢšĘĮ―éÓÚŲ|ÁŋÐÔ îšÍĩÁŋÐÔ îÖŪégĩÄŌŧîÐÔ îĢŽąíŽFéÓÐÐōĩÄîeĢŽĩŦß@ÐĐîeÖŪégĩÄēîŪēŧĘĮßBĀmĩÄĄĢąČČįēĄķūĩÖŋđÐÔËŪÆ―ĢĻļߥĒÖÐĄĒĩÍĢĐ/ŨŅÁĢîÉŦĢĻ\üSĄĒÉîüSĄĒŨØÉŦĩČĢĐ/ÖēÖęļßķČžeĢĻļߥĒÖÐĄĒĩÍĢĐĢŽß@ÐĐÐÔ îŋÉŌÔÓÃĩŨÖÖĩĢĻ1ĄĒ2ĄĒ3ĩČĢĐíÃčĘöĄĢ
ÄĮąíÐÍÎÄžþéLÉķÓÄØĢŋß@ĀïŌÔģĢŌžēēĄŅÐūŋéĀýĢŽÎÄžþŌŧ°ã°üšŽ3ÁÐĢšÓąūĢĻSampleĢĐĄĒÐÔeĢĻSexĢК͹íÐÍĢĻPhenotypeĢĐĄĢ
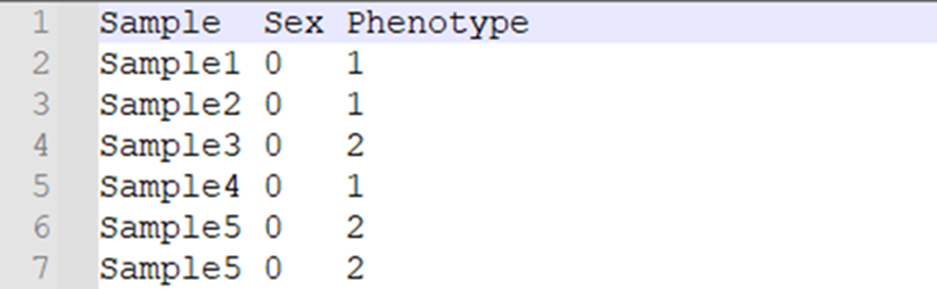
ŨĒŌâĢšÐÔeÓÃĩÖĩąíĘūĢŽ0ąíĘūÎīÖŠĄĒ1ąíĘūÄÐĄĒ2ąíĘūÅŪĄĒ-9ąíĘūȹʧĢŧąíÐÍŌēÓÃĩÖĩąíĘūĢŽ0ąíĘūÎīÖŠĄĒ1ąíĘūĶÕÕ―MĄĒ2ąíĘūō―MĄĒ-9ąíĘūȹʧĄĢēŧŌŠģöŽFŋÕČąÖĩĢĄ
02 ŧųŌōÐÍĩþ
ŧųŌōÐÍĘĮÖļŌŧówÔÚÄģĖØķĻŧųŌōÎŧücÉÏËųíÓÐĩÄĩČÎŧŧųŌōĩÄ―MšÏĄĢÃŋŧųŌōÎŧücŋÉŌÔÓÐēŧÍŽĩÄĩČÎŧŧųŌōĢŽß@ÐĐĩČÎŧŧųŌōĘĮÓÉļļÄļļũŨÔũßfĩÄŌŧÎąķów―MģÉĄĢ
ÄĮŧųŌōÐÍĩþÄÄÄĀïŦ@ČĄÄØĢŋÍĻģĢÓÐÉ·N·―Ę―ŋÉŌÔŦ@ČĄĢš
ĒŲ ŧųÓÚSNPÐūÆŽŦ@ČĄŧųŌōÐÍĩþ
ĒÚ ŧųÓÚŧųŌō―MyÐōŦ@ČĄŧųŌōÐÍĩþ
ß@ĀïÎŌŌÔVCFÎÄžþéĀýĢŽÄ”#CHROM”ÁÐé_ĘžūÍĘĮŨŪĩþĢš
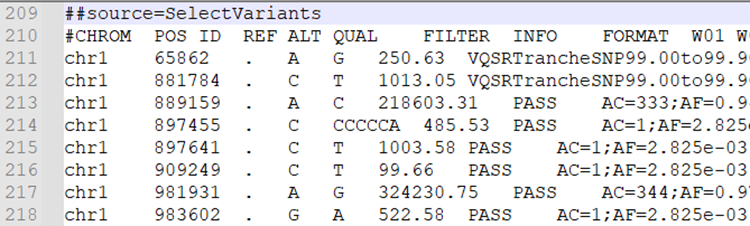
ŨĒŌâĢšÓÉÓÚÔĘžŨŪÎÄžþÖÐÓÐīóÁŋēŧŋÉŋŋŨŪĢŽ―ĻŨhĘđÓÃß^VšóĩÄVCFÎÄžþĢĻąČČįšYßxPASSĢĐßMÐКóĀm·ÖÎöĄĢ
ÓÉÓÚGWAS·ÖÎöÐčŌŠÓÝmŨ°ŲÉÏĮ§ĀýĩÄĩþĢŽŋÍôšÜëyýRÄĮÃīķāÓąūĢŽĖØeßŌŠî~ÍâĩÄÕýģĢÓąūŨũéĶÕÕĢŽĶŋÍôķøŅÔÓÖĘĮŌŧđPšÜīóĩÄÖ§ģöĢĄß@ršōÎŌŋÉŌÔēÉÓÞČÓÐĩÄÕýģĢČËĩþŨũéĶÕÕ―MĢŽŨîĩäÐÍĩÄūÍĘĮĘđÓÃĮ§ČËŧųŌō―MÓĩþéĶÕÕĄĢ
ŽFÔÚąíÐÍĩþšÍŧųŌōÐÍĩþķžÓÐÁËĢŽÔČįšÎĘđÓÃÄØĢŋÓÉÓÚÕûęP·ÖÎöÁũģĖĘĮŧųÓÚPLINKÜžþĢŽÄĮÃīūÍÐčŌŠĒŧųŌōÐÍĩþÞDQéPLINKÜžþÄÜŨReĩÄļņĘ―ĢŽÍŽrŌēĀûÓÚĖáļßĩþĖĀíЧÂĘĄĢPLINKÖÐÓÐÉîĩþļņĘ―ĢŽÔÚ·ÖÎöß^ģĖķžþÓÃĩ―Ģš
1. ped/mapļņĘ―
> pedļņĘ―ÎÄžþĢš
°üšŽÃŋÓąūĩÄŧųąūÐÅÏĒĢĻŨåÏĩĄĒÃû·QĄĒÐÔeĩČĢĐĄĒąíŽFÐÍÐÅÏĒĢĻphenotypeĢĐĄĒŧųŌōÐÍÐÅÏĒĢĻĩÚ7ÁÐÖŪšóĢĐĄĢÃŋÐÐąíĘūŌŧÓąūĢŽÔÎÄžþ]ÓÐąíî^ĢŽūßówÁÐĩČĄQÓÚÔÓąūËųšŽSNPsÎŧücĩĄĢ
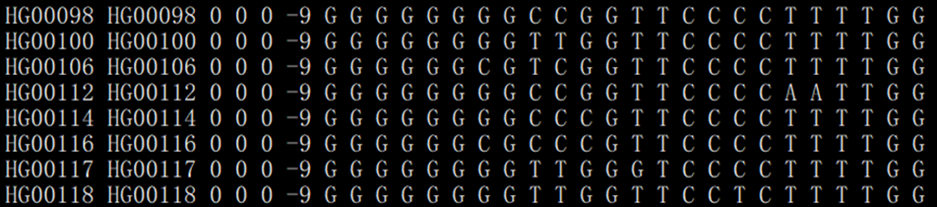
ĩÚŌŧÁÐĢšFamily IDĢŽŨåÏĩIDĢŧ
ĩÚķþÁÐĢšIndividual IDĢŽówĢĻČįÓąūégoŨåÏĩÂÏĩĢŽFamily IDšÍIndividual IDŋÉŌÔŌŧÓĢĐĢŧ
ĩÚČýÁÐĢšPaternal IDĢŽļļÏĩIDĢŽ0ąíĘūÎīÖŠĢŽ-1ąíĘūoļļÓH/ȹʧĢŧ
ĩÚËÄÁÐĢšMaternal IDĢŽÄļÏĩIDĢŽ0ąíĘūÎīÖŠĢŽ-1ąíĘūoÄļÓH/ȹʧĢŧ
ĩÚÎåÁÐĢšSexĢŽÐÔeĢŽ1 ÄÐĢŽ2 ÅŪĢŽ0 ŧō ÆäËûÖĩ ąíĘūÎīÖŠĢŧ
ĩÚÁųÁÐĢšPhenotypeĢŽąíÐÍÖĩĢŽļųþŅÐūŋîÐÍķĻÁxĢŧ
šóĀmÁÐĢšÄĩÚÆßÁÐé_ĘžĢŽÃŋÉÁÐīúąíÔÓąūËųšŽĩÄŌŧSNPĩÄŧųŌōÐÍĢšČįĩÚÆߥĒ°ËÁÐīúąíĩÚŌŧŧųŌōÐÍĢĻGGĢĐĢŽĩÚūÅĄĒĘŪÁÐīúąíĩÚķþŧųŌōÐÍĢĻGGĢĐĩČĩČĄĢ
> mapļņĘ―ÎÄžþĢš
ÓäÃŋ SNP ĩÄČūÉŦówÎŧÖÚÍßzũWÐÅÏĒĄĢÃŋÐÐĶŠŌŧ SNPĢŽÔÎÄžþ]ÓÐąíî^ĢŽÃŋÐаüšŽËÄÁÐĄĢ
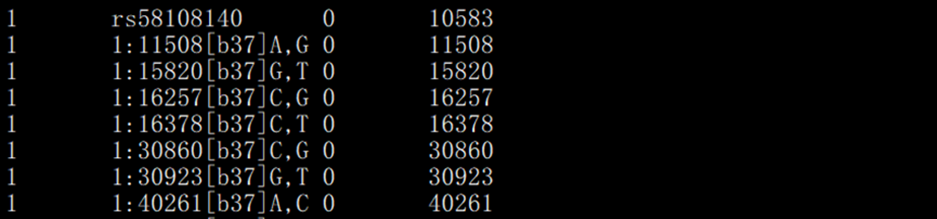
ĩÚŌŧÁÐĢšČūÉŦówūĖĢŧ
ĩÚķþÁÐĢšŨŪËŨR·ûĢŽß@ĀïĘĮrsūĖĢĻÓÐĩÄÔĢĐŧōÆäËû·―Ę―Ģŧ
ĩÚČýÁÐĢšßzũūāëxĢĻÄĶ ļųĢŽÎÎŧcMĢĐĢŽÎīÖŠĮérÏÂ0žīŋÉĢŧ
ĩÚËÄÁÐĢĻŋÉßxĢĐĢšSNPÔÚČūÉŦówÉÏÎïĀíÎŧÖÃĢŽļúĩÚČýÁÐąØÓÐŌŧÁÐĄĢ
2. bed/bim/famļņĘ―
> bedļņĘ―ÎÄžþĢš
īæĶŧųŌōÐÍÐÅÏĒĢĻķþßMÖÆĢĐĢŽÃŋÐÐĶŠŌŧÓąūĄĢÓÉÓÚĘĮķþßMÖÆļņĘ―ēŧÄÜÖą―Óīōé_ĄĢ
ĩÚŌŧÁÐĢšÓąūĩÄ IDĢĻÐčÅc .fam ÎÄžþÖÐĩÄ IID ŌŧÖÂĢĐĢŧ
šóĀmÁÐĢšÃŋÉÁÐéŌŧ SNP ĩÄÉĩČÎŧŧųŌōĢĻ°īíÐōÅÅÁÐĢŽČį A/C ŧō 0/1ĢĐĄĢÆäÖÐ0ĄĒ1ĄĒ2·ÖeĶŠÁËaaĄĒAaŧōaAšÍAAĄĢ
> binļņĘ―ÎÄžþĢš
īæĶÃŋ SNP ĩÄÔŠÐÅÏĒĢĻČūÉŦówÎŧÖÃĄĒßzũWūāëxĩČĢĐĄĢÃŋÐÐĶŠŌŧSNPĄĢ
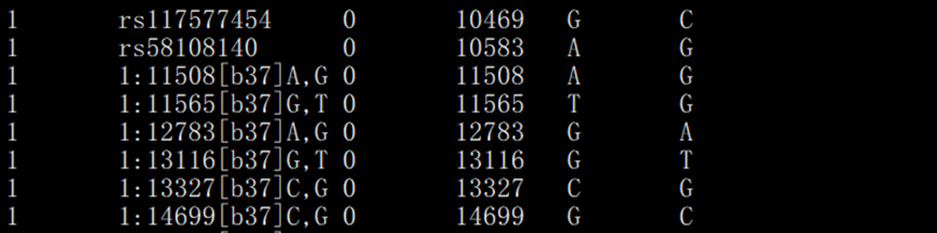
ĩÚŌŧÁÐĢšChrĢŽČūÉŦówūĖĢŧ
ĩÚķþÁÐĢšSNPĢŽËÓÃû·QĢŧ
ĩÚČýÁÐĢšGDĢŽßzũūāëxĢĻÄĶ ļųĢĐĢŽÎīÖŠĮérÏÂ0Ģŧ
ĩÚËÄÁÐĢšBPPĢŽŨŪÎŧücÎïĀíÎŧÖÃĢĻÎÎŧĢšbpĢĐĢŧ
ĩÚÎåÁÐĢšAllele 1ĢŽŌŧ°ãĮérÏÂéīÎŌŠĩČÎŧŧųŌōĢŧ
ĩÚÁųÁÐĢšAllele 2ĢŽŌŧ°ãĮérÏÂéÖũŌŠĩČÎŧŧųŌōĄĢ
> famļņĘ―ÎÄžþĢš
īæĶÓąūĩÄžŌÏĩęPÏĩšÍąíÐÍÐÅÏĒĢŽÃŋÐÐĶŠŌŧÓąūĄĢ

ĩÚŌŧÁÐĢšFIDĢŽžŌÏĩIDĢŧ
ĩÚķþÁÐĢšIIDĢŽówIDĢŧ
ĩÚČýÁÐĢšļļąūówIDĢŽ]ÓÐÓÃ0ąíĘūĢŽ-1ąíĘūȹʧĢŧ
ĩÚËÄÁÐĢšÄļąūówIDĢŽ]ÓÐÓÃ0ąíĘūĢŽ-1ąíĘūȹʧĢŧ
ĩÚÎåÁÐĢšSEXĢŽÐÔeĢŽ1ąíĘūÄÐÐÔĢŽ2ąíĘūÅŪÐÔĢŽ0ąíĘūÎīÖŠĢŧ
ĩÚÁųÁÐĢšPhenotypeĢŽąíÐÍÖĩĄĢ
ÐĄ―YĢšfamÎÄžþūÍĘĮpedÎÄžþĮ°ÁųÁÐĄĢīËÍâß@ĀïŋÉŌÔÍĻß^ĘÖÓĖížÓŅaČŦąíÐÍĄĒÐÔeÐÅÏĒĄĢ
ÄĮÔõÃīÍĻß^vcfļņĘ―ĩþĩÃĩ―ped/mapļņĘ―šÍbed/bim/famļņĘ―ĩþÄØĢŋÖą―ÓÉÏÃüÁîĢš
vcfÞDped/mapļņĘ―Ģš
plink --vcf test.vcf.gz --recode --out test
vcfÞDbed/bim/famļņĘ―Ģš
plink --vcf test.vcf.gz --make-bed --out test
ped/mapÞDbed/bim/famļņĘ―Ģš
plink --file test --make-bed --out test
bed/bim/famÞDped/mapļņĘ―Ģš
plink --bfile test --recode --out test
ŌÔÉÏūÍĘĮąūÆÚ·ÖÏíĩÄČČÝĢŽÏÂŌŧÆÚÎŌĒÖv―âČįšÎĶplinkļņĘ―ĩþßMÐÐŲ|ŋØß^VĄĢ

- ÉúÎïđŦđēĩþėĘđÓÃÖÐĩÄģĢŌî}šÍ―âQ·―°ļ
- ComplexHeatmap°üĀLÖÆÃÜķČáDĩÄ ĒĩÕ{ÕûšÍĖížÓŨĒá·―·Ļ
- DSPÅcLCMÖúÁĶ―âÎöŨÓ°BĮ°ÆÚÍÄĪŧŊĩÖŋđCÖÆ
- GWASČŦŧųŌō―MęP·ÖÎöĩÚŌŧÆÚĢšĩþž°ļņĘ―ÞDQ
- AIōÓūŦĘÄ[ÁöWÍŧÆÆĢšMUSKÄĢÐÍÖúÁĶátWŅÐūŋ
- ÔÚΞ°ûRNAyÐōÖÐDoubletFinderëpž°û·ÖÎö·―·ĻÔ―â
- scMegaÖúÁĶscATAC-seqĩþÅcscRNA-seqĩþĩÄšϷÖÎö
- ·ĮØūØę·Ö―âNMFËã·ĻÖúÁĶΞ°ûÞDä―Mĩþ·ÖÎö
- BSIŅûÄú ĒžÓĩ°°ŨŲ|―MWĩþ·ÖÎöÅāÓþ
- Ξ°ûŋÕégķā―MWžžÐgÕŊôßVÖÝÕūÉúÐÅÅāÓ°āóÃûÖÐ
- SBCΞ°ûž°ŋÕégķā―MWōÅcÉúÐÅ·ÖÎöÅāÓ°āÕÐÉú
- ÐĄšĢý°lēž°ŲMÏōÕnî}ĄĒĮ§ÆŠSCI-ÉúÃüŋÆWšÏŨũÓ
- ŋÕégķā―MWŅÐūŋēßÂÔž°ÉúÐÅ·ÖÎöÅāÓ°āŧðáÕÐÉúÖÐ
- SBC ToolBoxÔÆÆ―Å_VIPĢ ^ÔŲĖíÐÂÄĢKGSEAĩþ·ÖÎö
- SBCÐūÔÆÖvŊĢšøžŌŨÔČŧŋÆWŧų―ðĩÄĘäšÍÉęóžžĮÉ
- ĩÚĘŪÆÚSBCΞ°ûž°ŋÕégÞDä―MyÐōÅāÓ°āóÃûÍĻÖŠ