GWASČĢģųŌōŊMęP(gu¨Ąn)Â(li¨ĸn)ˇÖÎöĩÚŌģÆÚŖēĩ(sh¨´)ū(j¨´)ŧ°¸ņĘŊŪD(zhu¨Ŗn)Q
GWASŖ¨ČĢģųŌōŊMęP(gu¨Ąn)Â(li¨ĸn)ŅĐžŋŖŦGenome-Wide Association StudyŖŠĘĮŌģˇNͨß^ßÃčČĢģųŌōŊMˇļúČ(n¨¨i)ĩÄßz÷׎Ŗ¨ČįÎēËÜÕËáļāB(t¨¤i)ĐÔŖŦSNPŖŠŖŦ¤ÕŌÅcĖØļ¨ĐÔ îģōŧ˛˛Ąī@ÖøęP(gu¨Ąn)Â(li¨ĸn)ĩÄģųŌōÎģüc(di¨Ŗn)ĩġŊˇ¨ĄŖ
ÔÚßM(j¨Ŧn)ĐĐGWASˇÖÎöÖŽĮ°ŖŦĐčŌĒßxņēĪßmĩÄČēķwŖŦßM(j¨Ŧn)ĐĐĩ(sh¨´)ū(j¨´)ĘÕŧ¯ĄŖ(sh¨Ē)ëHš¤×÷ÖĐ°l(f¨Ą)ŦF(xi¨¤n)ĶĐĐŠŋÍô¸ųąž˛ģÖĒĩĀGWASˇÖÎöĐčŌĒĘ˛Ã´ĩ(sh¨´)ū(j¨´)ŖŦĩĸÕ`ˇÖÎößM(j¨Ŧn)ļČŖŦÄĮĐčŌĒĘÕŧ¯ÄÄĐŠĩ(sh¨´)ū(j¨´)ÄØŖŋÖ÷ŌĒĐčŌĒÉ(g¨¨)ĩ(sh¨´)ū(j¨´)ŖēąíĐÍĩ(sh¨´)ū(j¨´)ēÍģųŌōĐÍĩ(sh¨´)ū(j¨´)ĄŖ
01 ąíĐÍĩ(sh¨´)ū(j¨´)
ÕÆđąíĐÍĩ(sh¨´)ū(j¨´)ŖŦÎŌĪČíÁËŊâĪÂGWASĩÄąíĐÍĐÔ îĄŖGWASÖĐĩÄąíĐÍĐÔ îŋÉŌÔˇÖéŌÔĪÂČũîŖē
ĸŲ ĩ(sh¨´)ÁŋĐÔ îŖ¨Quantitative TraitsŖŠŖēĘĮÖ¸ŋÉŌÔĶÃĩ(sh¨´)×ÖÖĩíÃčĘöĩÄĐÔ îĄŖąČČįÉí¸ßŖ¨cmŖŠ/ķwÖØŖ¨kgŖŠ/×ŅÁŖĩ(sh¨´)Ŗ¨(g¨¨)ŖŠ/Ža(ch¨Ŗn)ÁŋŖ¨kg/ŽŖŠŖŦˇÖÎör(sh¨Ē)ąíĐÍÖąŊĶĘšĶÞßķwĩ(sh¨´)ÖĩŖģ
ĸÚ Ų|(zh¨Ŧ)ÁŋĐÔ îŖ¨Qualitative TraitsŖŠŖēÅcĩ(sh¨´)ÁŋĐÔ îĪāˇ´ŖŦÆäoˇ¨ĶÚĖļ¨ĩ(sh¨´)ÖĩąíĘžŖŦļøĘĮąíŦF(xi¨¤n)ŗöŌģˇN îB(t¨¤i)ĄŖąČČįģ¨ÉĢŖ¨ŧtĄĸüSĄĸ°×ĩČŖŠ/šû(sh¨Ē)ĐÎ îŖ¨AĄĸEAĩČŖŠ/ŧ˛˛ĄŖ¨ĶĐģōoŖŠŖŦąíĐÍŋÉŌÔĶÃĩ(sh¨´)Öĩē(ji¨Ŗn)ģ¯ąíĘžŖģ
ĸÛ ˇÖŧ(j¨Ē)ĐÔ îŖ¨Ordinal TraitsŖŠŖēĘĮŊéĶÚŲ|(zh¨Ŧ)ÁŋĐÔ îēÍĩ(sh¨´)ÁŋĐÔ îÖŽégĩÄŌģîĐÔ îŖŦąíŦF(xi¨¤n)éĶĐĐōĩÄîeŖŦĩĢß@ĐŠîeÖŽégĩIJģĘĮßBĀm(x¨´)ĩÄĄŖąČČį˛ĄļžĩÖŋšĐÔËŽÆŊŖ¨¸ßĄĸÖĐĄĸĩÍŖŠ/×ŅÁŖîÉĢŖ¨\üSĄĸÉîüSĄĸ×ØÉĢĩČŖŠ/Ö˛Öę¸ßļČŧ(j¨Ē)eŖ¨¸ßĄĸÖĐĄĸĩÍŖŠŖŦß@ĐŠĐÔ îŋÉŌÔĶÃĩ(sh¨´)×ÖÖĩŖ¨1Ąĸ2Ąĸ3ĩČŖŠíÃčĘöĄŖ
ÄĮąíĐÍÎÄŧūéL(zh¨Ŗng)ÉļĶÄØŖŋß@ĀīŌÔŗŖŌŧ˛˛ĄŅĐžŋéĀũŖŦÎÄŧūŌģ°ã°üēŦ3ÁĐŖēĶąžŖ¨SampleŖŠĄĸĐÔeŖ¨SexŖŠēÍąíĐÍŖ¨PhenotypeŖŠĄŖ
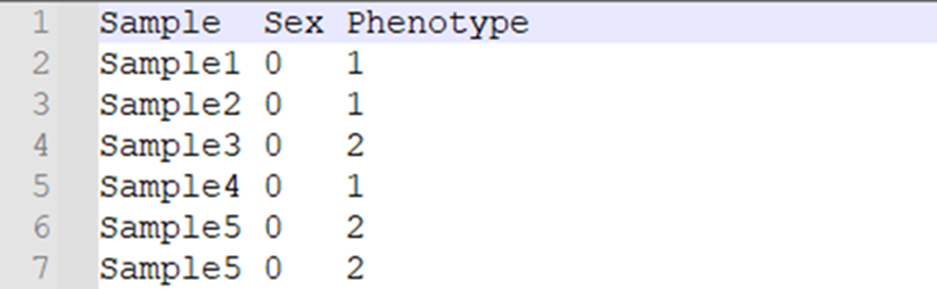
×ĸŌâŖēĐÔeĶÃĩ(sh¨´)ÖĩąíĘžŖŦ0ąíʞδÖĒĄĸ1ąíĘžÄĐĄĸ2ąíĘžÅŽĄĸ-9ąíʞȹʧŖģąíĐÍŌ˛ĶÃĩ(sh¨´)ÖĩąíĘžŖŦ0ąíʞδÖĒĄĸ1ąíĘžĻ(du¨Ŧ)ÕÕŊMĄĸ2ąíĘž(sh¨Ē)ō(y¨¤n)ŊMĄĸ-9ąíʞȹʧĄŖ˛ģŌĒŗöŦF(xi¨¤n)ŋÕČąÖĩŖĄ
02 ģųŌōĐÍĩ(sh¨´)ū(j¨´)
ģųŌōĐÍĘĮÖ¸Ōģ(g¨¨)(g¨¨)ķwÔÚÄŗ(g¨¨)ĖØļ¨ģųŌōÎģüc(di¨Ŗn)ÉĪËųíĶĐĩÄĩČÎģģųŌōĩÄŊMēĪĄŖÃŋ(g¨¨)ģųŌōÎģüc(di¨Ŗn)ŋÉŌÔĶвģÍŦĩÄĩČÎģģųŌōŖŦß@ĐŠĩČÎģģųŌōĘĮĶɸ¸Ä¸¸÷×Ô÷ßfĩÄŌģ(g¨¨)ÎąļķwŊMŗÉĄŖ
ÄĮģųŌōĐÍĩ(sh¨´)ū(j¨´)ÄÄÄĀīĢ@ČĄÄØŖŋͨŗŖĶĐɡNˇŊĘŊŋÉŌÔĢ@ČĄŖē
ĸŲ ģųĶÚSNPĐžÆŦĢ@ČĄģųŌōĐÍĩ(sh¨´)ū(j¨´)
ĸÚ ģųĶÚģųŌōŊMy(c¨¨)ĐōĢ@ČĄģųŌōĐÍĩ(sh¨´)ū(j¨´)
ß@ĀīÎŌŌÔVCFÎÄŧūéĀũŖŦÄ”#CHROM”ÁĐé_ĘŧžÍĘĮ׎ĩ(sh¨´)ū(j¨´)Ŗē
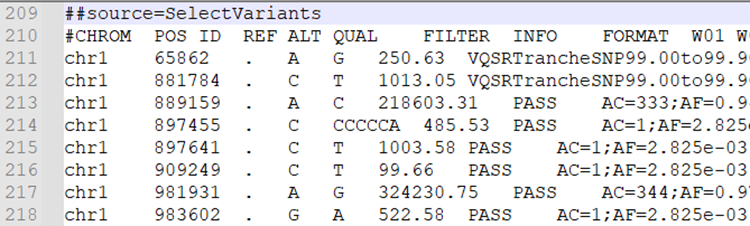
×ĸŌâŖēĶÉĶÚÔĘŧ׎ÎÄŧūÖĐĶĐ´ķÁŋ˛ģŋÉŋŋ׎ŖŦŊ¨×hĘšĶÃß^VēķĩÄVCFÎÄŧūŖ¨ąČČįēYßxPASSŖŠßM(j¨Ŧn)ĐĐēķĀm(x¨´)ˇÖÎöĄŖ
ĶÉĶÚGWASˇÖÎöĐčŌĒ?ji¨Ŗng)ĶŨm×°ŲÉĪĮ§ĀũĩÄĩ(sh¨´)ū(j¨´)ŖŦŋÍôēÜëyũRÄĮôļāĶąžŖŦĖØeßŌĒî~ÍâĩÄÕũŗŖĶąž×÷éĻ(du¨Ŧ)ÕÕŖŦĻ(du¨Ŧ)ŋÍôļøŅÔĶÖĘĮŌģšPēÜ´ķĩÄÖ§ŗöŖĄß@(g¨¨)r(sh¨Ē)ēōÎŌŋÉŌÔ˛ÉĶÃŧČĶĐĩÄÕũŗŖČËĩ(sh¨´)ū(j¨´)×÷éĻ(du¨Ŧ)ÕÕŊMŖŦ×îĩäĐÍĩÄžÍĘĮĘšĶÃĮ§ČËģųŌōŊMĶ(j¨Ŧ)ĩ(sh¨´)ū(j¨´)éĻ(du¨Ŧ)ÕÕĄŖ
ŦF(xi¨¤n)ÔÚąíĐÍĩ(sh¨´)ū(j¨´)ēÍģųŌōĐÍĩ(sh¨´)ū(j¨´)ļŧĶĐÁËŖŦÔČįēÎĘšĶÃÄØŖŋĶÉĶÚÕû(g¨¨)ęP(gu¨Ąn)Â(li¨ĸn)ˇÖÎöÁ÷ŗĖĘĮģųĶÚPLINKÜŧūŖŦÄĮôžÍĐčŌĒĸģųŌōĐÍĩ(sh¨´)ū(j¨´)ŪD(zhu¨Ŗn)QéPLINKÜŧūÄÜ×R(sh¨Ē)eĩĸņĘŊŖŦÍŦr(sh¨Ē)Ō˛ĀûĶÚĖá¸ßĩ(sh¨´)ū(j¨´)ĖĀíЧÂĘĄŖPLINKÖĐĶĐÉîĩ(sh¨´)ū(j¨´)¸ņĘŊŖŦÔÚˇÖÎöß^ŗĖļŧū(hu¨Ŧ)ĶÃĩŊŖē
1. ped/map¸ņĘŊ
> ped¸ņĘŊÎÄŧūŖē
°üēŦÃŋ(g¨¨)ĶąžĩÄģųąžĐÅĪĸŖ¨×åĪĩĄĸÃûˇQĄĸĐÔeĩČŖŠĄĸąíŦF(xi¨¤n)ĐÍĐÅĪĸŖ¨phenotypeŖŠĄĸģųŌōĐÍĐÅĪĸŖ¨ĩÚ7ÁĐÖŽēķŖŠĄŖÃŋĐĐąíĘžŌģ(g¨¨)ĶąžŖŦÔÎÄŧū]ĶĐąíî^ŖŦžßķwÁĐĩ(sh¨´)ČĄQĶÚÔĶąžËųēŦSNPsÎģüc(di¨Ŗn)ĩ(sh¨´)ĄŖ
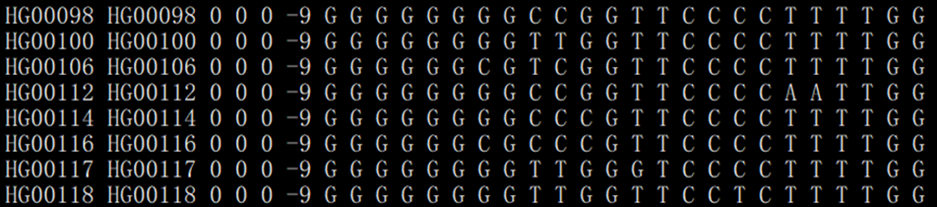
ĩÚŌģÁĐŖēFamily IDŖŦ×åĪĩIDŖģ
ĩÚļūÁĐŖēIndividual IDŖŦ(g¨¨)ķwŖ¨ČįĶąžégo×åĪĩÂ(li¨ĸn)ĪĩŖŦF(xi¨¤n)amily IDēÍIndividual IDŋÉŌÔŌģĶŖŠŖģ
ĩÚČũÁĐŖēPaternal IDŖŦ¸¸ĪĩIDŖŦ0ąíʞδÖĒŖŦ-1ąíĘžo¸¸ĶH/ȹʧŖģ
ĩÚËÄÁĐŖēMaternal IDŖŦĸĪĩIDŖŦ0ąíʞδÖĒŖŦ-1ąíĘžoĸĶH/ȹʧŖģ
ĩÚÎåÁĐŖēSexŖŦĐÔeŖŦ1 ÄĐŖŦ2 ÅŽŖŦ0 ģō ÆäËûÖĩ ąíʞδÖĒŖģ
ĩÚÁųÁĐŖēPhenotypeŖŦąíĐÍÖĩŖŦ¸ųū(j¨´)ŅĐžŋîĐÍļ¨ÁxŖģ
ēķĀm(x¨´)ÁĐŖēÄĩÚÆßÁĐé_ĘŧŖŦÃŋ?j¨Š)ÉÁĐ´úąíÔĶąžËųēŦĩÄŌģ(g¨¨)SNPĩÄģųŌōĐÍŖēČįĩÚÆߥĸ°ËÁĐ´úąíĩÚŌģ(g¨¨)ģųŌōĐÍŖ¨GGŖŠŖŦĩÚžÅĄĸĘŽÁĐ´úąíĩÚļū(g¨¨)ģųŌōĐÍŖ¨GGŖŠĩČĩČĄŖ
> map¸ņĘŊÎÄŧūŖē
ĶäÃŋ(g¨¨) SNP ĩÄČžÉĢķwÎģÖÃēÍßz÷W(xu¨Ļ)ĐÅĪĸĄŖÃŋĐĐĻ(du¨Ŧ)Ē(y¨Šng)Ōģ(g¨¨) SNPŖŦÔÎÄŧū]ĶĐąíî^ŖŦÃŋĐĐ°üēŦËÄÁĐĄŖ
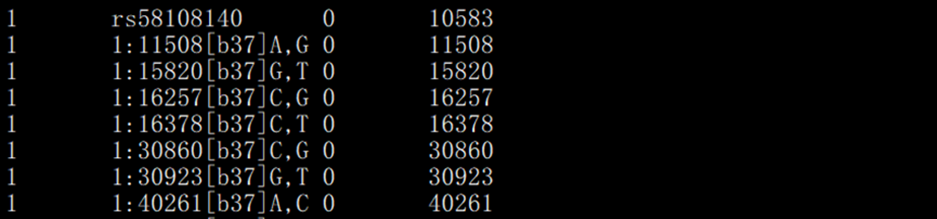
ĩÚŌģÁĐŖēČžÉĢķwžĖ(h¨¤o)Ŗģ
ĩÚļūÁĐŖē׎Ë(bi¨Ąo)×R(sh¨Ē)ˇûŖŦß@ĀīĘĮrsžĖ(h¨¤o)Ŗ¨ĶĐĩÄÔŖŠģōÆäËûˇŊĘŊŖģ
ĩÚČũÁĐŖēßz÷žāëxŖ¨ÄĻ ¸ųŖŦÎÎģcMŖŠŖŦδÖĒĮérĪÂ0ŧ´ŋÉŖģ
ĩÚËÄÁĐŖ¨ŋÉßxŖŠŖēSNPÔÚČžÉĢķwÉĪÎīĀíÎģÖÃŖŦ¸úĩÚČũÁĐąØĶĐŌģÁĐĄŖ
2. bed/bim/fam¸ņĘŊ
> bed¸ņĘŊÎÄŧūŖē
´æĻ(ch¨ŗ)ģųŌōĐÍĐÅĪĸŖ¨ļūßM(j¨Ŧn)ÖÆŖŠŖŦÃŋĐĐĻ(du¨Ŧ)Ē(y¨Šng)Ōģ(g¨¨)ĶąžĄŖĶÉĶÚĘĮļūßM(j¨Ŧn)ÖƸņĘŊ˛ģÄÜÖąŊĶ´ōé_ĄŖ
ĩÚŌģÁĐŖēĶąžĩÄ IDŖ¨ĐčÅc .fam ÎÄŧūÖĐĩÄ IID ŌģÖÂŖŠŖģ
ēķĀm(x¨´)ÁĐŖēÃŋ?j¨Š)ÉÁĐéŌģ(g¨¨) SNP ĩÄÉ(g¨¨)ĩČÎģģųŌōŖ¨°´íĐōÅÅÁĐŖŦČį A/C ģō 0/1ŖŠĄŖÆäÖĐ0Ąĸ1Ąĸ2ˇÖeĻ(du¨Ŧ)Ē(y¨Šng)ÁËaaĄĸAaģōaAēÍAAĄŖ
> bin¸ņĘŊÎÄŧūŖē
´æĻ(ch¨ŗ)Ãŋ(g¨¨) SNP ĩÄÔĒĐÅĪĸŖ¨ČžÉĢķwÎģÖÃĄĸßz÷W(xu¨Ļ)žāëxĩČŖŠĄŖÃŋĐĐĻ(du¨Ŧ)Ē(y¨Šng)Ōģ(g¨¨)SNPĄŖ
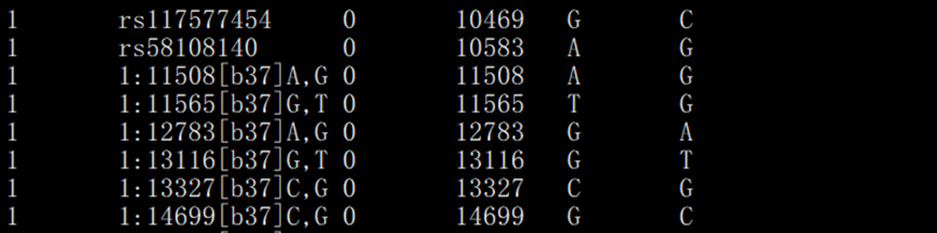
ĩÚŌģÁĐŖēChrŖŦČžÉĢķwžĖ(h¨¤o)Ŗģ
ĩÚļūÁĐŖēSNPŖŦË(bi¨Ąo)ĶÃûˇQŖģ
ĩÚČũÁĐŖēGDŖŦßz÷žāëxŖ¨ÄĻ ¸ųŖŠŖŦδÖĒĮérĪÂ0Ŗģ
ĩÚËÄÁĐŖēBPPŖŦ׎Îģüc(di¨Ŗn)ÎīĀíÎģÖÃŖ¨ÎÎģŖēbpŖŠŖģ
ĩÚÎåÁĐŖēAllele 1ŖŦŌģ°ãĮérĪÂé´ÎŌĒĩČÎģģųŌōŖģ
ĩÚÁųÁĐŖēAllele 2ŖŦŌģ°ãĮérĪÂéÖ÷ŌĒĩČÎģģųŌōĄŖ
> fam¸ņĘŊÎÄŧūŖē
´æĻ(ch¨ŗ)ĶąžĩÄŧŌĪĩęP(gu¨Ąn)ĪĩēÍąíĐÍĐÅĪĸŖŦÃŋĐĐĻ(du¨Ŧ)Ē(y¨Šng)Ōģ(g¨¨)ĶąžĄŖ

ĩÚŌģÁĐŖēFIDŖŦŧŌĪĩIDŖģ
ĩÚļūÁĐŖēIIDŖŦ(g¨¨)ķwIDŖģ
ĩÚČũÁĐŖē¸¸ąž(g¨¨)ķwIDŖŦ]ĶĐĶÃ0ąíĘžŖŦ-1ąíʞȹʧŖģ
ĩÚËÄÁĐŖēĸąž(g¨¨)ķwIDŖŦ]ĶĐĶÃ0ąíĘžŖŦ-1ąíʞȹʧŖģ
ĩÚÎåÁĐŖēSEXŖŦĐÔeŖŦ1ąíĘžÄĐĐÔŖŦ2ąíĘžÅŽĐÔŖŦ0ąíʞδÖĒŖģ
ĩÚÁųÁĐŖēPhenotypeŖŦąíĐÍÖĩĄŖ
ĐĄŊY(ji¨Ļ)ŖēfamÎÄŧūžÍĘĮpedÎÄŧūĮ°ÁųÁĐĄŖ´ËÍâß@ĀīŋÉŌÔͨß^ĘÖĶ(d¨°ng)ĖíŧĶŅa(b¨ŗ)ČĢąíĐÍĄĸĐÔeĐÅĪĸĄŖ
ÄĮÔõôͨß^vcf¸ņĘŊĩ(sh¨´)ū(j¨´)ĩÃĩŊped/map¸ņĘŊēÍbed/bim/fam¸ņĘŊĩ(sh¨´)ū(j¨´)ÄØŖŋÖąŊĶÉĪÃüÁîŖē
vcfŪD(zhu¨Ŗn)ped/map¸ņĘŊŖē
plink --vcf test.vcf.gz --recode --out test
vcfŪD(zhu¨Ŗn)bed/bim/fam¸ņĘŊŖē
plink --vcf test.vcf.gz --make-bed --out test
ped/mapŪD(zhu¨Ŗn)bed/bim/fam¸ņĘŊŖē
plink --file test --make-bed --out test
bed/bim/famŪD(zhu¨Ŗn)ped/map¸ņĘŊŖē
plink --bfile test --recode --out test
ŌÔÉĪžÍĘĮąžÆÚˇÖĪíĩÄČ(n¨¨i)ČŨŖŦĪÂŌģÆÚÎŌĸÖvŊâČįēÎĻ(du¨Ŧ)plink¸ņĘŊĩ(sh¨´)ū(j¨´)ßM(j¨Ŧn)ĐĐŲ|(zh¨Ŧ)ŋØß^VĄŖ

- ÉúÎīšĢš˛ĩ(sh¨´)ū(j¨´)ėĘšĶÃÖĐĩÄŗŖŌî}ēÍŊâQˇŊ°¸
- ComplexHeatmap°üĀLÖÆÃÜļČáDĩÄ ĸĩ(sh¨´)Õ{(di¨¤o)ÕûēÍĖíŧĶ×ĸáˇŊˇ¨
- DSPÅcLCMÖúÁĻŊâÎö×Ķ°BĮ°ÆÚÍĤģ¯ĩÖŋšC(j¨Š)ÖÆ
- GWASČĢģųŌōŊMęP(gu¨Ąn)Â(li¨ĸn)ˇÖÎöĩÚŌģÆÚŖēĩ(sh¨´)ū(j¨´)ŧ°¸ņĘŊŪD(zhu¨Ŗn)Q
- AIō(q¨ą)Ķ(d¨°ng)žĢĘ(zh¨ŗn)Ä[ÁöW(xu¨Ļ)ÍģÆÆŖēMUSKÄŖĐÍÖúÁĻát(y¨Š)W(xu¨Ļ)ŅĐžŋ
- ÔÚÎŧ(x¨Ŧ)°ûRNAy(c¨¨)ĐōÖĐDoubletFinderëpŧ(x¨Ŧ)°ûˇÖÎöˇŊˇ¨ÔŊâ
- scMegaÖúÁĻscATAC-seqĩ(sh¨´)ū(j¨´)ÅcscRNA-seqĩ(sh¨´)ū(j¨´)ĩÄÂ(li¨ĸn)ēĪˇÖÎö
- ˇĮØ(f¨´)žØęˇÖŊâNMFËãˇ¨ÖúÁĻÎŧ(x¨Ŧ)°ûŪD(zhu¨Ŗn)äŊMĩ(sh¨´)ū(j¨´)ˇÖÎö
- BSIŅûÄú ĸŧĶĩ°°×Ų|(zh¨Ŧ)ŊMW(xu¨Ļ)ĩ(sh¨´)ū(j¨´)ˇÖÎöÅāĶ(x¨´n)ū(hu¨Ŧ)
- Îŧ(x¨Ŧ)°ûŋÕégļāŊMW(xu¨Ļ)ŧŧĐg(sh¨´)Õ¯ôßVÖŨÕžÉúĐÅÅāĶ(x¨´n)°āķ(b¨¤o)ÃûÖĐ
- SBCÎŧ(x¨Ŧ)°ûŧ°ŋÕégļāŊMW(xu¨Ļ)(sh¨Ē)ō(y¨¤n)ÅcÉúĐÅˇÖÎöÅāĶ(x¨´n)°āÕĐÉú
- ĐĄēŖũ°l(f¨Ą)˛ŧ°Ų(g¨¨)MĪōÕnî}ĄĸĮ§ÆĒSCI-ÉúÃüŋÆW(xu¨Ļ)ēĪ×÷Ķ(j¨Ŧ)
- ŋÕégļāŊMW(xu¨Ļ)ŅĐžŋ˛ßÂÔŧ°ÉúĐÅˇÖÎöÅāĶ(x¨´n)°āģđáÕĐÉúÖĐ
- SBC ToolBoxÔÆÆŊÅ_(t¨ĸi)VIPŖ ^(q¨ą)ÔŲĖíĐÂÄŖKGSEAĩ(sh¨´)ū(j¨´)ˇÖÎö
- SBCĐžÔÆÖv¯Ŗēø(gu¨Ž)ŧŌ×ÔČģŋÆW(xu¨Ļ)ģųŊđĩÄĘ(zh¨ŗn)äēÍÉęķ(b¨¤o)ŧŧĮÉ
- ĩÚĘŽÆÚSBCÎŧ(x¨Ŧ)°ûŧ°ŋÕégŪD(zhu¨Ŗn)äŊMy(c¨¨)ĐōÅāĶ(x¨´n)°āķ(b¨¤o)ÃûͨÖĒ