
De Novo輔助的DIA同源序列檢索提升低豐度突變肽的鑒定
數據非依賴采集(DIA)可通過劃分采集窗口,將所有檢測范圍內的離子進行碎裂和掃描,定量準確性高、重現性好、蛋白質組覆蓋深度廣,尤其適合低豐度肽段的檢測。但現有DIA數據分析方法主要依賴譜圖庫或直接數據庫搜索,但兩者均受限于數據庫中的已知肽段列表,難以發現由遺傳變異或突變產生的新生肽段。2025年5月30日,Bioinformatics Solutions Inc.發表了最新預印本文章 [1],介紹了一種直接從復雜的DIA譜圖中發現低豐度突變肽段的算法,同時嚴格控制FDR。
DIAVariants工作流程
如圖1所示,讀取數據后,首先進行MS1特征峰提取,以識別電離肽段產生的具有明確同位素的可靠MS1信號響應。接下來進行PEAKS DIA數據庫搜索,將譜圖與參考蛋白質序列庫比對,篩選FDR小于1%的可信肽段,并基于保留時間和母離子 m/z與MS1特征峰關聯。
對數據庫匹配不可信的MS1特征峰,利用DIA de novo預測與關聯MS2最佳匹配的肽段序列。然后通過SPIDER算法,基于de novo預測的序列、MS1特征峰信息和參考序列校正測序錯誤。SPIDER候選肽段中,僅保留包含1-2個突變位點的序列,下一步預測這些候選突變肽的索引保留時間(iRT),通過RT回歸模型轉換為實驗RT,過濾掉預測iRT與實測RT差異顯著的突變肽段。最后,將候選突變肽與數據庫搜索得到的肽段合并構建臨時譜圖庫,基于實測RT進行PEAKS DIA譜圖庫搜索,通過Q值控制FDR。
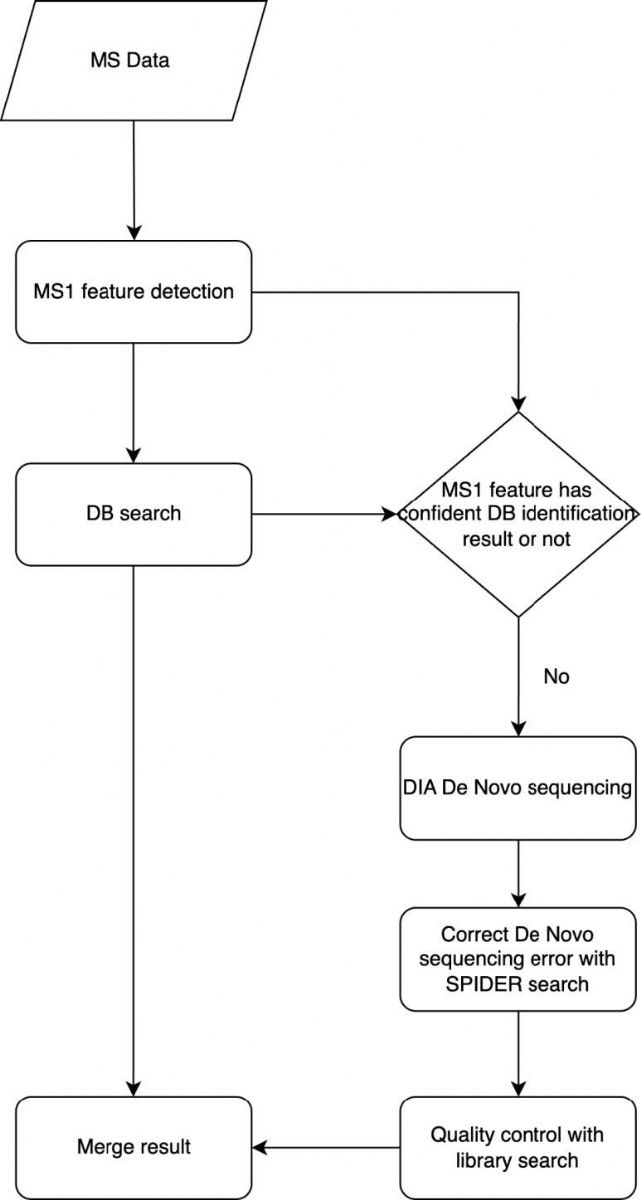
圖1 DIAVariant方法流程
結果展示PEAKS DIA直接數據庫搜索性能
下載兩個公開質譜數據集(PXD046453、PXD050030),采用相同的database和參數,分別使用PEAKS DIA DB search和DIANN2.1進行分析,結果如表1所示,從定量蛋白數量、CV指標來看,兩者在DIA蛋白質組數據的定量性能上,穩定性和靈敏度均較高。
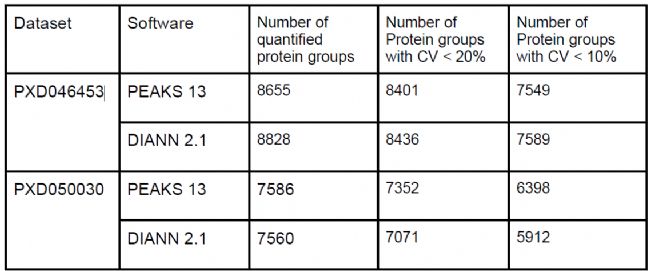
表1 文獻數據分析結果對比
跨物種搜索驗證DIAVariants特異性
在ABRF人類樣本(搜索小鼠數據庫)和MSV000095360小鼠樣本(搜索人類數據庫)的結果中,DIAVariant報告的肽段變體中85%以上可在本物種數據庫中被可信鑒定(q-value <0.01),驗證了算法的高特異性(表2)。
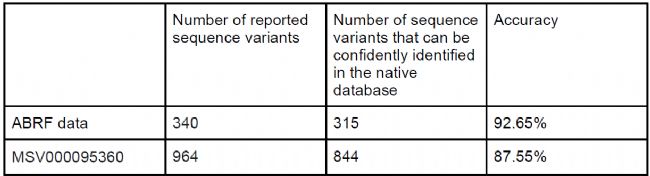
表2 跨物種檢索結果
與蛋白基因組方法對比
Fierro-Monti等[2]曾對Hela細胞樣本進行外顯子組測序,通過蛋白基因組的方法構建了233個經典蛋白變體,將其加入人類參考序列數據庫后,使用DIANN 1.8.1檢索到了相應的DIA數據,并通過同位素合成肽段和靶向驗證,最終報告了6個肽段變體。我們下載了文獻中的原始數據,用DIAVariants直接分析,結果成功鑒定到了上述6條肽段變體的其中2條(LEQDLQQIQAK 和 NELSGALTGLIR)(表 3),并且額外發現426個低豐度潛在多肽變體。這些低豐度肽段因MS1信號弱,難以通過DDA數據檢測,更加顯示了DIA-MS在低豐度肽段檢測中的優勢。
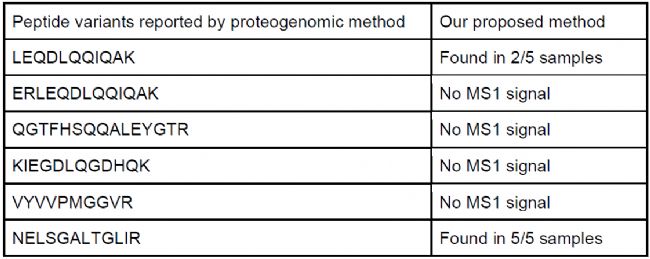
表3 DIAVariants與蛋白基因組報道肽段對比。
小結DIAVariant通過整合DIA數據庫搜索、從頭測序和同源校正,提供了一種高效、無偏的DIA數據突變肽段識別方法,可同時檢測參考數據庫內的已知肽段和數據庫外的肽段變體,尤其適用于低豐度肽段和復雜遺傳變異的分析。該方法為蛋白質組學中序列變體的發現提供了新工具,有望推動精準醫學和癌癥新抗原研究的發展。但目前僅驗證了1-2個氨基酸突變位點,未涉及插入和缺失的情況,未來算法仍會繼續擴展。
參考文獻
[1] Qiao, R., et al. (2025). "De Novo sequencing-assisted homology search for DIA data analysis enables low abundance peptide variants discovery." doi: https://doi.org/10.1101/2025.05.30.657054.
[2] Fierro-Monti, Ivo, et al. "Assessment of Data-Independent Acquisition Mass Spectrometry (DIA-MS) for the Identification of Single Amino Acid Variants." Proteomes 12.4 (2024): 33.
原文鏈接:https://www.biorxiv.org/content/10.1101/2025.05.30.657054v1



Copyright(C) 1998-2025 生物器材網 電話:021-64166852;13621656896 E-mail:info@bio-equip.com