阿爾茨海默病腦血管系統(tǒng)的單核多區(qū)轉(zhuǎn)錄組分析的應(yīng)用

期刊:Nature Neuroscience
影響因子:25
技術(shù)服務(wù):單細胞核測序
導語
腦血管失調(diào)是阿爾茨海默病(AD)的一個標志,但發(fā)生在特定細胞類型的變化尚未完全確定。本文分析了220名AD患者和208名年齡匹配的對照者的6個大腦區(qū)域的人類腦血管系統(tǒng)中的snRNA-seq。注釋了22,514個腦血管細胞,包括內(nèi)皮細胞、周細胞、平滑肌細胞、血管周圍成纖維細胞和室管膜細胞。鑒定出2676個AD差異表達基因,包括周細胞中PDGFRB的下調(diào),內(nèi)皮細胞中ABCB1和ATP10A的下調(diào),并驗證了死后AD腦組織中SLC6A1的下調(diào)和APOD、INSR和COL4A1的上調(diào)。檢測到血管、神經(jīng)膠質(zhì)和神經(jīng)元共表達的基因模塊,提示AD中神經(jīng)血管單位的協(xié)調(diào)失調(diào)。
與AD遺傳學的整合揭示了125個AD差異表達基因與AD相關(guān)的遺傳變異直接相關(guān)。最后,發(fā)現(xiàn)APOE4基因型相關(guān)差異在毛細血管和小靜脈內(nèi)皮細胞以及周細胞和成纖維細胞亞群中的AD相關(guān)基因中顯著富集。
技術(shù)方法
snRNA-seq
研究結(jié)果
1. 橫跨六個腦區(qū)的腦血管特征
對阿爾茨海默病(AD)患者和對照組的六個大腦區(qū)域的腦血管進行了snRNA-seq,以發(fā)現(xiàn)人類腦血管細胞及其轉(zhuǎn)錄組差異。共檢測了22,514個單個核的轉(zhuǎn)錄組,并注釋了11種血管細胞類型,包括內(nèi)皮細胞、周細胞、平滑肌細胞、血管周細胞和成纖維細胞等。還發(fā)現(xiàn)了在動脈、毛細血管和靜脈內(nèi)皮細胞、動脈和靜脈平滑肌細胞之間具有不同的轉(zhuǎn)錄組特征,并指出了它們在不同血管類型中的特殊功能。
預測了與分子功能和細胞身份相關(guān)活動的上游調(diào)節(jié)因子。發(fā)現(xiàn)主要細胞類型共享上游調(diào)節(jié)因子,但仍然表現(xiàn)出亞型特異性。一些基因在血管內(nèi)皮細胞中高度富集,如CTNNB1,它通過內(nèi)皮β-catenin信號通路維持BBB的完整性。對于PER,發(fā)現(xiàn)BACH1的富集,這與它通過調(diào)節(jié)血管生成素-1的表達來進行轉(zhuǎn)錄調(diào)控是一致的。
接下來評估了血管細胞在大腦區(qū)域和表型變量之間是否存在豐度差異。發(fā)現(xiàn)在內(nèi)嗅皮層、海馬和丘腦中FIBs的比例顯著較高,PERs和cEndo細胞的比例顯著較低,這與大腦中小血管的缺乏一致。與這些區(qū)域差異相反,血管細胞組分并不隨性別、AD病理、年齡或死亡時間間隔(PMI)而不同。檢測到的基因數(shù)量或捕獲的細胞數(shù)量與細胞比例之間沒有顯著的相關(guān)性。然而,與血管富集數(shù)據(jù)集相比,海馬和PFC中FIBs (FIB1和FIB2)的比例明顯更高、,這表明捕獲血管周圍樣FIBs可能對所使用的技術(shù)敏感。
此外,我們發(fā)現(xiàn)血管細胞在腦區(qū)之間表現(xiàn)出廣泛的基因表達差異,其中1,745個基因在兩個腦區(qū)之間存在差異表達。

Fig 1. 橫跨六個腦區(qū)的腦血管特征
2. AD患者細胞類型特異性腦血管改變
為了研究血管基因表達與AD之間的關(guān)系,基于單細胞的方法結(jié)合不同腦區(qū)域所有細胞類型的細胞,在AD患者和對照組之間鑒定了2676個AD DEGs(adDEGs)。通過排列分析,adDEGs的數(shù)量顯著高于的預期,并在pseudobulk水平得到證。其中,2142個僅為一種細胞類型所特有。值得注意的是,cEndo細胞具有最高數(shù)量的adDEGs,cEndo功能中轉(zhuǎn)錄差異的重要性與AD病理相關(guān)。
其中,細胞連接相關(guān)和粘附相關(guān)基因,包括APOD、PECAM1和COLEC12;包括SLC38A2、SLC2A1和SLC6A1在內(nèi)的轉(zhuǎn)運蛋白,以及與固醇輸入相關(guān)的基因,包括RORA、PRKAA2和PPARG,表明血管細胞類型的這些基本功能可能是AD 中可能存在失調(diào)。正如預期的那樣, AD樣本的PERs中PDGFRB的陽性率,暗示PERAD1,13中BBB完整性的損傷和功能障礙。ABCB1基因在cEndo細胞中明顯下調(diào)。細胞數(shù)量最多的三種細胞類型(cEndo,PER1和FIB1)顯示出最大的重疊。
對adDEGs進行GO富集分析表明,多種廣泛的功能途徑在細胞類型之間是共享的,這表明AD中的轉(zhuǎn)錄變化在通路水平上富集到共同的功能;然而,許多途徑更具細胞類型特異性。
CD31+血管片段在個體AD中具有較高的INSR1轉(zhuǎn)錄密度。發(fā)現(xiàn)AD大腦中CD31+內(nèi)皮細胞的子集具有較高密度的INSR+。這些表明內(nèi)皮細胞的不同亞群可能表達不同水平的胰島素受體,使細胞對胰島素信號的敏感性增強或減弱。編碼高密度脂蛋白(HDL)成分的APOD在AD壁細胞中上調(diào),這與以前報道的APOD在AD中的上調(diào)一致。為了驗證AD PER中APOD的上調(diào),使用原位雜交定量GRM8標記細胞中的APOD轉(zhuǎn)錄豐度,并觀察到在患有AD的個體中GRM8+ PERs具有較高的APOD表達。PER中APOD上調(diào)的功能后果以及對神經(jīng)血管單元內(nèi)脂質(zhì)轉(zhuǎn)運的影響將成為未來研究的基礎(chǔ)。此外,驗證了SLC6A1在AD PER中的下調(diào),以及COL4A1在AD的血管內(nèi)皮細胞中的上調(diào)。

Fig 2. AD中細胞類型特異性腦血管改變
3. AD中DEGs的上游調(diào)控因子
鑒定了118個上調(diào)adDEG的上游調(diào)節(jié)因子和81個下調(diào)adDEG的上游調(diào)節(jié)因子,其中66個靶向各種細胞類型中上調(diào)和下調(diào)的adDEG。這133個調(diào)節(jié)因子中有17個是相應(yīng)的差異表達基因。當這些調(diào)節(jié)因子顯示出目標基因的顯著共享時,定義為共調(diào)節(jié)模塊(定義為tfModules)。對于cEndo中上調(diào)的adDEG,鑒定了7個tfModule,包括38個調(diào)節(jié)因子,以及11個不在tfModules中的調(diào)節(jié)因子。同一tfModule中的調(diào)節(jié)因子通常具有相關(guān)功能。 在每個tfModule中,調(diào)節(jié)因子具有各自靶基因數(shù)量、在AD中的差異表達以及靶基因的差異表達。并非每個tfModule中的所有調(diào)節(jié)因子都是DEG,這表明一些調(diào)節(jié)因子可能通過與差異表達的調(diào)節(jié)因子的協(xié)作發(fā)揮作用。
使用這些tfModules將目標adDEG分為由不同tfModule組合介導的亞組。對于cEndo上調(diào)的adDEG,發(fā)現(xiàn)由單一tfModule靶向的群體和其他由多個tfModules靶向的群體,具有獨特的共同功能富集。相反,G1中的adDEG是由大多數(shù)tfModules(M1-M4、M6)的調(diào)節(jié)因子靶向的,并顯著富集在細胞因子反應(yīng)、細胞粘附和轉(zhuǎn)錄調(diào)控中。幾個群體中的基因(G3、G7、G8)富集在抑制內(nèi)皮細胞增殖和凋亡信號通路的表達上,這表明在AD中存在內(nèi)皮細胞損傷反應(yīng),這與BBB破壞一致。

Fig 3. AD中DEGs的上游調(diào)控因子
4. AD細胞間通訊動力學
通過分析跨越409個個體的基因模塊(定義為degModules)的協(xié)變分析,預測了細胞之間的雙向通信。發(fā)現(xiàn)了301個在AD中上調(diào)的相互作用,其中相互作用的degModules在AD中均顯示上調(diào)。來自PER1和cEndo細胞到神經(jīng)元、小膠質(zhì)細胞和星形膠質(zhì)細胞的通信在AD中占主導地位,而來自星形膠質(zhì)細胞和神經(jīng)元到cEndo細胞和FIB1s的通信在AD中主要減少。
為了表征這種差異細胞間通信的配體-受體信號通路,將由相同配體-受體對介導的單個相互作用聚集起來。TGFβ、SPP1、BMP、ANGPTL和IL-6介導的信號通路在AD增加的相互作用中顯著高表達,而膠原蛋白和層粘連蛋白是形成BBB基底膜的主要細胞外基質(zhì)(ECM)蛋白,其在AD減少的相互作用中高表達,這表明AD中BBB結(jié)構(gòu)被破壞。
排列測試發(fā)現(xiàn)大多數(shù)雙向細胞間相互作用的變化在AD中每種細胞類型對之間都是顯著的,這表明AD中失調(diào)的基因與多細胞相互作用高度相關(guān)。接下來,將每個細胞類型對之間的雙向AD差異細胞間相互作用分為正向和反向方向,并對它們進行了量化。cEndo和PER1細胞以及興奮性神經(jīng)元和星形膠質(zhì)細胞之間的相互作用最多。為三個細胞-細胞對(cEndo-Ex、cEndo-Astro和PER1-Ex)構(gòu)建了配體-受體網(wǎng)絡(luò),以突出特定的配體-受體信號通路。BMP6介導PERs(PER1)和興奮性神經(jīng)元(Ex)之間AD增加的相互作用,表明其在PER中控制神經(jīng)發(fā)生受損的作用。星形膠質(zhì)細胞和cEndo細胞之間由表皮生長因子(EGF)信號通路介導的AD減少的相互作用表明,在AD中抑制了cEndo的增殖。

Fig 4. AD全基因組關(guān)聯(lián)研究基因座與腦血管病相關(guān)
5. AD全基因組關(guān)聯(lián)研究基因座與腦血管病相關(guān)
將AD相關(guān)的位點與adDEG整合在一起,以預測其候選靶基因和作用方向,其作用的細胞類型以及它們可以直接或間接導致血管基因表達變化的機制。197個AD相關(guān)變異位于113個位點,它們接近125個血管adDEG,顯示出細胞類型特異性表達變化。大多數(shù)adDEG在其內(nèi)含子(54.3%)或緊鄰上游或下游且無介入基因(7.13%)中攜帶AD相關(guān)變異,或者根據(jù)不同證據(jù)相互關(guān)聯(lián)。這些AD相關(guān)的adDEG富集在多個過程中,包括膽固醇運輸、調(diào)節(jié)內(nèi)皮細胞遷移和IL-6介導的信號。21個GWAS位點與脂質(zhì)和膽固醇代謝adDEG基因相關(guān)聯(lián),這與AD中廣泛失調(diào)的腦膽固醇穩(wěn)態(tài)是一致的。
9個AD GWAS位點與免疫反應(yīng)、胰島素分泌和神經(jīng)退行性病變有關(guān),包括IL6和IL6R,它們在AD患者的cEndo細胞中高表達,提示內(nèi)皮細胞的免疫反應(yīng)可能是AD血管的一個突出特征。
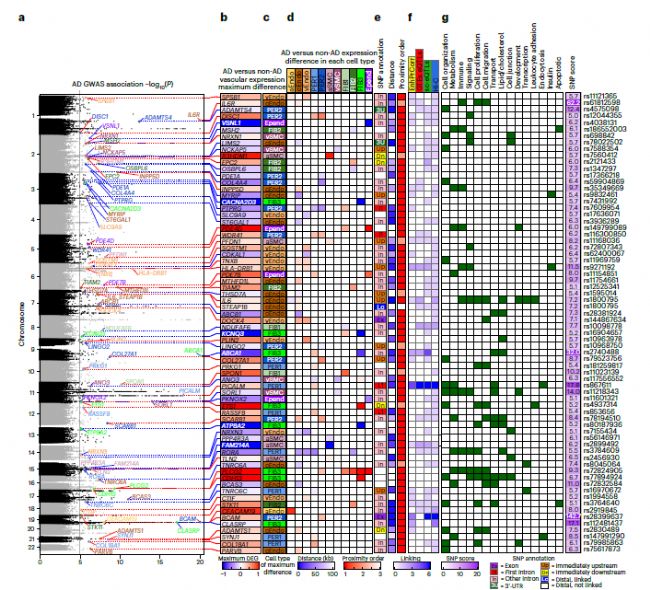
Fig 5. AD全基因組關(guān)聯(lián)研究基因座與腦血管病相關(guān)
尋找間接遺傳證據(jù)以證明血管adDEG的上游調(diào)節(jié)器直接與AD相關(guān)變異(轉(zhuǎn)錄調(diào)控)相關(guān)聯(lián)。使用了之前注釋的相關(guān)模塊對(degModules),并搜索了AD相關(guān)遺傳位點的adDEG配體,以尋找潛在的遺傳效應(yīng)。發(fā)現(xiàn)了54對(24個上調(diào)和30個下調(diào)在AD中),其配體靠近AD相關(guān)遺傳位點,涉及611個與12個AD相關(guān)配體和13個受體(18個不同的配體-受體對)相關(guān)聯(lián)的血管adDEG。這12個AD相關(guān)配體代表了比預期更顯著的富集。其中7個配體顯示了單細胞表達數(shù)量性狀位點(sc-eQTL)連接證據(jù),3個顯示了組織水平腦eQTL證據(jù),7個顯示了Hi-C環(huán)連接證據(jù)。其下游基因至少富集在17種不同的生物學功能中。總之,使用順式、轉(zhuǎn)錄或細胞間信號調(diào)節(jié)機制,AD中的2676個血管adDEG中有1010個可以與AD遺傳關(guān)聯(lián)。這表明遺傳風險因素對腦血管的影響可能有助于AD的發(fā)病機制,這是通過腦血管細胞類型和大腦實質(zhì)神經(jīng)、膠質(zhì)和巨噬細胞以及外周血中的免疫細胞的細胞內(nèi)功能障礙和細胞間通訊實現(xiàn)的。

Fig 6. AD GWAS基因與腦血管adDEGs間接相關(guān)
6. APOE基因型與血管細胞認知功能下降
2482個apoeDEG在細胞類型中的分布大體均衡(平均而言,120個APOE4上調(diào)和120個APOE4下調(diào)),并且數(shù)量與adDEG相似。雖然只有中位數(shù)為4%的APOE差異基因與AD差異基因重疊,但apoeDEG與adDEG之間的重疊對于一些細胞類型來說具有重要意義。對于cEndo細胞,36%的apoeDEG上調(diào)基因也是adDEG上調(diào)基因,21%的apoeDEG下調(diào)基因也是adDEG下調(diào)基因。FIB1和PER1細胞中發(fā)現(xiàn)了強有力的apoeDEG-adDEG一致性(十倍至十四倍富集),而PER2和vEndo上調(diào)基因的一致性較弱(六倍至八倍)。
搜索了每個細胞類型中上調(diào)和下調(diào)的apoeDEG所富集的生物學途徑。在cEndo細胞中,下調(diào)的apoeDEG(E4-down)顯著富集在跨BBB運輸、細胞連接組織以及調(diào)節(jié)芽生血管生成中。在PERs中,下調(diào)的apoeDEG富集在細胞遷移調(diào)節(jié)、跨BBB運輸以及細胞-細胞連接維持中,這與APOE ε4個體表現(xiàn)出腦血流量減少和腦血管異常增加的報道一致。評估了所有apoeDEG與認知衰退的相關(guān)性,發(fā)現(xiàn)上調(diào)的apoeDEG(E4-up)與所有細胞類型的認知衰退相關(guān),而下調(diào)的apoeDEG(E4-down)主要與認知恢復相關(guān)。這種效果在cEndo、vEndo、FIB1和PER1細胞中最為明顯,E4-up和E4-down基因在認知衰退相關(guān)性方面存在實質(zhì)性和高度顯著差異。結(jié)果與之前的研究結(jié)果一致,cPERs可能介導APOE4對認知衰退的影響,并表明cEndo和FIB1細胞可能具有同等甚至更重要的作用。
進一步鑒定了APOE基因型條件性的、與認知衰退相關(guān)的差異表達基因(cogDEG),區(qū)分了‘認知衰退上調(diào)’cogDEG和‘認知衰退下調(diào)’cogDEG。前者與認知衰退呈正相關(guān),后者與認知衰退呈負相關(guān)。APOE4個體比APOE3個體表現(xiàn)出更多的cogDEG(超過1.5倍),特別是在cEndo、vEndo、PER1和FIB1細胞中,尤其對于認知衰退上調(diào)基因(2.1倍),這表明特定的腦血管細胞類型可能介導APOE ε4等位基因?qū)φJ知衰退的貢獻。比較APOE3與APOE4 cogDEG,發(fā)現(xiàn)這兩組基因大部分不同,只有約5%的cogDEG在APOE4和APOE3之間是共同的,這表明ε3和ε4攜帶者在轉(zhuǎn)錄變化和認知衰退的具體機制上存在差異。
為了進一步研究APOE基因型條件性的cogDEG的功能,在毛細血管細胞類型(cEndo和PER1)中對E3特異性的、E4特異性的和E3-E4共享的cogDEG進行了GO富集分析。對于cEndo認知衰退上調(diào)的cogDEG,APOE4特異性富集包括脂質(zhì)和細胞因子反應(yīng)以及細胞凋亡過程;APOE3富集包括血管運輸、細胞遷移的負調(diào)節(jié)和細胞基質(zhì)粘附。對于cEndo認知衰退下調(diào)的cogDEG,APOE4特異性富集包括血管發(fā)育、BMP信號的調(diào)節(jié)和神經(jīng)遞質(zhì)轉(zhuǎn)運;APOE3富集包括內(nèi)皮細胞遷移的正調(diào)節(jié)和細胞外結(jié)構(gòu)組織。對于PER1認知衰退上調(diào)的cogDEG,APOE4特異性富集包括細胞因子反應(yīng)、細胞-細胞連接組裝和細胞凋亡;APOE3富集包括SMC遷移的負調(diào)節(jié)和Notch信號,以及自噬。對于PER1認知衰退下調(diào)的cogDEG,APOE4特異性富集包括BBB相關(guān)功能(BBB維持、Notch信號、收縮調(diào)節(jié)),而APOE3富集包括脂質(zhì)和化學穩(wěn)態(tài)以及細胞連接組裝,這表明APOE ε4依賴的認知衰退可能主要由PER1 PERs介導。

Fig 7. APOE4-相關(guān)的轉(zhuǎn)錄變化和認知能力下降
參考文獻:
Sun N, Akay LA, Murdock MH, Park Y, Galiana-Melendez F, Bubnys A, Galani K, Mathys H, Jiang X, Ng AP, Bennett DA, Tsai LH, Kellis M. Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer's disease. Nat Neurosci. 2023 Jun;26(6):970-982. doi: 10.1038/s41593-023-01334-3. Epub 2023 Jun 1. Erratum in: Nat Neurosci. 2023 Oct 31;: PMID: 37264161; PMCID: PMC10464935.